
Синдром VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) — відкрита у 2020 році нозологія, яка об’єднує риси автозапальних та гематологічних хвороб. Від моменту першого опису минуло лише п’ять років, а Американський коледж ревматології разом із Міжнародною робочою групою VEXAS опублікували перші міждисциплінарні рекомендації з ідентифікації та лікування цього синдрому (2025). Документ також схвалений Міжнародним товариством системних автозапальних захворювань.
Етіологія і патогенез
Причина — соматичні мутації в гені UBA1 (Xp11.23), які порушують функцію ферменту E1 у системі убіквітина. Це веде до активації вродженого імунітету, хронічного запалення та формування гематологічних змін.
VEXAS не є спадковим захворюванням.
Епідеміологія
- Найчастіше хворіють чоловіки старше 50 років.
- Задокументовано близько 450 випадків, але глобальна поширеність може сягати 1 млн осіб.
- Патогенні варіанти UBA1 зустрічаються приблизно у 1 з 13 591 осіб.
Клінічні прояви
Широкий спектр симптомів ускладнює діагностику.
- Системні: персистуюча лихоманка, схуднення, високі показники CRP/ШОЕ.
- Запальні ознаки: вушний/носовий хондрит, висип, періорбітальний набряк, вузлики в легенях, венозний тромбоз.
- Шкіра: нейтрофільні дерматози (часто діагностують як Sweet-синдром).
- Очі: увеїт, склерит.
- Легені: інтерстиціальні інфільтрати.
- Суглоби/серце: артрит, міо- чи перикардит.
- Нирки: інтерстиціальний нефрит.
- Гематологічні ознаки:
- макроцитоз, макроцитарна анемія,
- тромбоцитопенія, моноцитопенія, лімфопенія,
- вакуолі у проеритробластах і промієлоцитах,
- часта асоціація з мієлодиспластичним синдромом (МДС) чи іншими гематологічними неоплазіями.
- макроцитоз, макроцитарна анемія,
Важливо: клінічна картина може варіюватися від тяжкого автозапального фенотипу до ізольованих цитопеній.
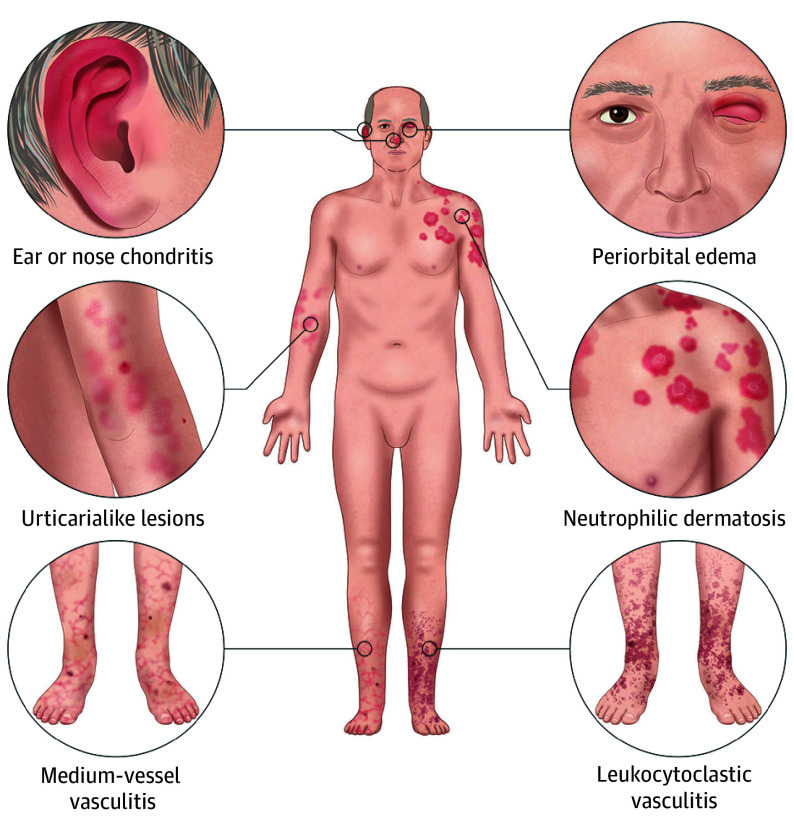
Діагностика
Міжнародні рекомендації 2025 року наголошують:
- Генетичне тестування:
- основний метод — NGS з аналізом екзону 3 гена UBA1;
- якщо тест негативний, слід секвенувати увесь ген або досліджувати кістковий мозок.
- основний метод — NGS з аналізом екзону 3 гена UBA1;
- Кістковий мозок:
- біопсія рекомендована при цитопеніях;
- складність інтерпретації через часту дисплазію, що не завжди відповідає класичним критеріям МДС.
- біопсія рекомендована при цитопеніях;
- Клінічна настороженість:
- чоловік >40 років, із рефрактерним системним запаленням, макроцитозом, цитопеніями та «незрозумілими» проявами з боку різних органів.
- чоловік >40 років, із рефрактерним системним запаленням, макроцитозом, цитопеніями та «незрозумілими» проявами з боку різних органів.
Лікування
Цілі терапії:
- контроль запалення,
- профілактика кістковомозкової недостатності,
- зменшення ускладнень від лікування,
- покращення якості життя.
Основні підходи:
- Глюкокортикоїди — базисна терапія, але висока залежність.
- JAK-інгібітори та IL-6-інгібітори — ефективніші за стандартні DMARDs та B-клітинні агенти (ритуксимаб).
- Гіпометилюючі препарати (азацитидин) — показані при поєднанні з МДС.
- Алогенна трансплантація стовбурових клітин — єдиний потенційно радикальний метод.
- Критично важливо контролювати ризики тромбозів та інфекцій (основні причини летальних випадків).
Мультидисциплінарний підхід
Пацієнти з VEXAS можуть звертатися до різних спеціалістів:
- гематологи — через цитопенії та МДС,
- ревматологи — через артрит, хондрит, системні запальні прояви,
- дерматологи — через поліморфні висипи,
- пульмонологи — при легеневих інфільтратах,
- судинні спеціалісти — при тромбозах,
- нефрологи — при ниркових ураженнях.
Саме тому нові рекомендації підкреслюють необхідність координації ведення в рамках мультидисциплінарної команди.
Прогноз
- 5-річна виживаність ~80%.
- Несприятливі фактори: мутація p.Met41Val, тромбоцитопенія <100 тис., трансфузійна залежність, супутні мутації DNMT3A/TET2.
Висновок
Синдром VEXAS — новий виклик для гематологів, ревматологів, нефрологів та інших спеціалістів. Він може імітувати різні хвороби, а без специфічної діагностики залишатися невизнаним.






